Frequenzberechnung
Die Änderung der potentiellen Energie $V$ der Kerne kann durch eine Taylor-Reihe um Entwicklungspunkt $X_0$ ausgedrückt werden:
\[V(X) = V(X_0) + g^T(X-X_0) + \frac{1}{2}(X-X_0)^T H (X-X_0) + ...\]wobei wiederum $g$ der Gradient und $H$ die Hesse-Matrix ist. Die harmonische Näherung vereinfacht diese Taylor-Reihe indem nur die Terme bis zur zweiten Ableitung berücksichtigt werden. Im Energie-Minimum ist der Gradient $g^T=0$ und mit der Wahl $V(X_0)=0$ ergibt sich:
\[V(X) = \frac{1}{2}(X-X_0)^T H (X-X_0)\]Die Elemente der Hesse-Matrix sind die zweiten Ableitungen der potentiellen Energie nach den Kernpositionen. Durch Diagonalisierung (Ähnlichkeitstransformation) wechselt man in das Koordinatensystem der Normalmoden und die Hesse-Matrix wird zur Diagonalmatrix $\Lambda$. Die Beiträge einzelner Atomkerne gehen hierbei massegewichtet in die Hesse-Matrix ein. Die Eigenwerte $\lambda_i$ entsprechen den Kraftkonstanten und können in Schwingungsfrequenzen $\nu_i$ umgerechnet werden:
\[\nu_i = \frac{1}{2\pi} \sqrt{\frac{\lambda_i}{\mu}}\]Wird diese harmonische Näherung korrekt auf eine Minimums-Geometrie angewandt so sind alle Eigenwerte reell und positiv.
Falls ein Sattelpunkt vorliegt sind einige Eigenwerte negativ und die entsprechenden Frequenzen imaginär.
Folgende Abbildung zeigt die ersten drei Normalmoden des Wassermoleküls. Diese entsprechen der antisymmetrischen- und symmetrischen Streckschwingung, sowie der Biegeschwingung.
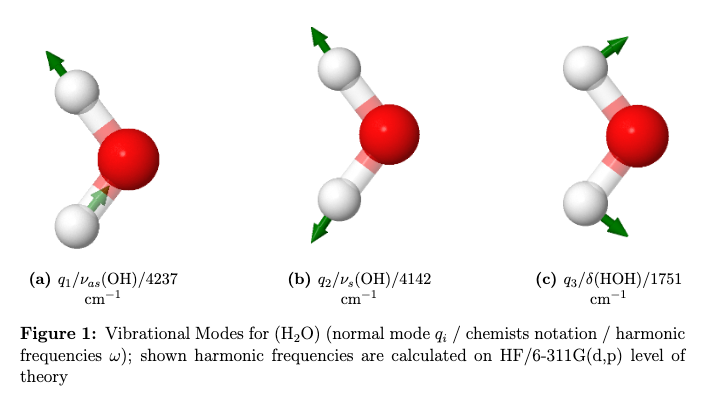
Frequenzberechnung in Orca
Folgender Input-File ist ein Beispiel für eine Frequenzberechnung in Orca. Als Methode wird wiederum Hartree Fock mit dem 6-311G(d,p) Basissatz verwendet.
!HF 6-311G(d,p) OPT FREQ
* xyz 0 1
H 0.000000000 1.415075762 0.956290882
O 0.000000000 0.000000000 -0.120510070
H 0.000000000 -1.415075762 0.956290882
*
Es wird sowohl eine Geometrie-Optimierung (OPT) als auch eine Frequenzberechnung (FREQ) durchgeführt. Grundsätzlich kann die Frequenzberechnung analytisch (wenn verfügbar) oder numerisch (durch z.B. Finite-Differenzen) durchführt werden.
-----------------------
VIBRATIONAL FREQUENCIES
-----------------------
Scaling factor for frequencies = 1.000000000 (already applied!)
0: 0.00 cm**-1
1: 0.00 cm**-1
2: 0.00 cm**-1
3: 0.00 cm**-1
4: 0.00 cm**-1
5: 0.00 cm**-1
6: 1750.79 cm**-1
7: 4141.74 cm**-1
8: 4237.00 cm**-1
------------
NORMAL MODES
------------
These modes are the Cartesian displacements weighted by the diagonal matrix
M(i,i)=1/sqrt(m[i]) where m[i] is the mass of the displaced atom
Thus, these vectors are normalized but *not* orthogonal
0 1 2 3 4 5
0 0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
1 0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
2 0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
3 0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
4 0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
5 0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
6 0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
7 0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
8 0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
6 7 8
0 -0.000000 -0.000000 -0.000000
1 -0.430096 -0.583118 -0.561293
2 0.559049 -0.398411 -0.427118
3 0.000000 0.000000 0.000000
4 -0.000000 0.000001 0.070729
5 -0.070445 0.050202 -0.000001
6 -0.000000 0.000000 0.000000
7 0.430096 0.583097 -0.561316
8 0.559049 -0.398396 0.427133
Im Output-File findet man dann die berechneten Frequenzen in $\text{cm}^{-1}$ und die zugehörigen massegewichteten Normalmoden.
Aufgabe 2a
Berechnen Sie die harmonischen Frequenzen für alle Moleküle ihrer Reaktion und vergleiche Sie diese mit experimentellen Daten. Machen Sie sich bewusst um welche Art von Experiment es sich handelt. Verwenden Sie für die Berechnung die folgenden Methoden-Basissatz Kombinationen:
- HF/6-311G(d,p)
- B3LYP/6-311G(d,p)
- MP2/6-311G(d,p)
Wichtig!!
In Orca sind für die MP2-Methode keine analytischen zweiten Ableitungen (Hesse-Matrix) implementiert. Daher muss die Hesse-Matrix numerisch über Finite-Differenzen berechnet werden. Dies kann durch die Angabe der Option numfreq im Input-File erreicht werden.
Plotten von Normalmoden
Zum Plotten und visualisieren der Normalmoden verwenden wir das Programm Jmol dieses ist unter folgenden Link kostenlos zum Download verfügbar:
https://jmol.sourceforge.net/
Will man die Vibrationen eines Moleküls also nach einer abgeschlossenen Frequenzrechung von Orca visualisieren geht man wie folgt vor:
- Man generiert Trajektorien zu den jeweiligen Moden mittels den Befehl
orca_pltvib jobname.out all. - Als Resultat erhält man Files vom Typ
jobname.out.v00x.xyzwelche man dann mit Jmol öffnen kann - Hierzu verwendet man den Befehl
jmol jobname.out.v00x.xyz. - Mittels des graphischen Interfaces von Jmol oder der zugehörigen Konsole kann man dann die Vektoren der Normalmoden anzeigen lassen und Bilder für die Präsentation erstellen.
Verständnisfragen
- Wie erkennt man anhand der harmonischen Schwingungsfrequenzen ob ein Energie-Minimum oder ein Sattelpunkt vorliegt?
- Was sind Normalmoden? Wie werden diese berechnet und konstruiert?
- Wie kommt man von den Eigenwerten der Hesse-Matrix zu den Schwingungsfrequenzen?
- Welche Möglichkeiten gibt es für anharmonische Korrekturen der Schwingungsfrequenzen?
- Warum unterscheiden sich harmonische Frequenzen so stark von den experimentellen Werten? Welche Effekte werden in der harmonischen Näherung nicht berücksichtigt?
- Wie viele Vibrationsmoden hat ein Molekül mit $N$ Atomen?
- Wie beurteilt man ob eine Schwingung IR-aktiv ist?
- Was sind Obertöne und Kombinationsbanden?